GSvit v 1.8
GSvit architecture is based on use of computational core (GSvit) and a GUI (XSvit). This
page provides documentation for the computational core, you can also check the
documentation for graphical user interface.
Installation
Please see the download page.
Running the computational core
To run a regular calculation,
GSvit program must be started with parameter file as a command line argument. If any other files are used
(e.g. to specify material or source) these are also mentioned in the parameter file. Parameter file
is an ASCII file composed by keywords and sets of values corresponding to these values. Rows can be
commented out using '#' character at the beginning of the row. Check examples
at end of this page to see some working parameter files.
Automated tests
If GSvit is started with "test" as an argument, followed by test number, it can run an automated
test. For key tests even a precomputed value is installed, so result can be compared to it automatically.
If a GPU is present in the computer and GSvit is translated with GPU support, it is recognized, reported
and used for comparison to CPU.
Tests can be useful namely when installing GSvit on a new system or in doubts whether a concrete
algorithm does not do what is expected.
Automated tests available are as follows (run gsvit with no arguments for a list of them):
- ./gsvit test 0 checks available GPUs only, printing their main characteristics.
- ./gsvit test 1 makes single test on CPU and all GPUs (if there are any)
- ./gsvit test 2 tests key algorithms performance, running approximately ten different tests for 100x100x100 voxels, 100 steps, which takes some ten minutes.
- ./gsvit test 3 tests key algorithms performance, running approximately ten different tests for 200x200x200 voxels, 300 steps, which can take tens of minutes, however
is more precise regarding peak GSvit performance estimation.
- ./gsvit test 12 compares GPU/CPU time scaling up to 200x200x200 voxels
- ./gsvit test 13 compares GPU/CPU time scaling up to 300x300x300 voxels
- ./gsvit test 14 compares GPU/CPU time scaling up to 400x400x400 voxels
- ./gsvit test 15 compares GPU/CPU time scaling up to 500x500x500 voxels
- ./gsvit test 20 compares GPU time scaling up to 400x400x400 voxels
- ./gsvit test 3N tests multiple threads speedup on N00xN00xN00 voxels (i.e. 100 to 900 for 31 to 39)
- ./gsvit test AAABCDEF is a general test format. The eigth digit number AAABCDEF means:
- AAA - cube size in pixels (i.e. if XXX = 100, cube size is 100 x 100 x 100)
- B - boundary condition: 0 - none, 1 - PEC, 2 - Liao, 3 - CPML
- C - material (small piece of it in the center of computational space): 0 - none, 1 - electric only, 2 - magnetic only, 3 - tabulated electric, 4 - tabulated magnetic, 5 - pec, 6 - Drude, 7 - CP
- D - check material and appl optimum mode for saving some memory: 0 - no, 1 - yes
- E - run Near-to Far Field transformation for a single point: 0 - no, 1 - yes
- F - source: 0 - point source, 1 - total/scattered field, 2 - scattered field
The "AAABCDEF" naming is used also to store the precomputed data that are compared with tests results. The precomputed
data for comparison are typically single near field or far field point time dependence.
There are two more tests designed for benchmarking HPC systems:
- ./gsvit benchmark N tests N threads computational time on 900x900x900 voxels
- ./gsvit bigbenchmark N tests N threads computational time on 4000x4000x4000 voxels
One more special command line argument is implemented. Parameter "--killer filename" defines
special condition for stopping calculation if the specified file in working directory exists
and its content (ASCII) is a number 1. This specific workaround could be useful only very
rarely, at present its main use is to prevent wrong process termination in Windows version
of XSvit.
Parameter file description
Main computation settings
Keywords are case sensitive. The following keywords can be used in the current version of GSvit (before issuing version 1.0 this means actual verison as obtained from SVN, but nearly all the parameters
remain same for last issued binaries):
POOL
xres yres zres dx dy dz
Set computation volume dimensions in pixels (xres, yres, zres) and real pixel size - pixel spacing (dx, dy, dz) in meters. To create a 200x200x200 pixel
computational volume with pixel spacing of 1 micrometer, set:
POOL
200 200 200 1e-6 1e-6 1e-6
COMP
nsteps
Set total number of computation steps, e.g. to set the number of steps to 100, write:
COMP
100
DT_MULT
value
Set multiplication factor for automated time step estimation - using value e.g. 0.5 shortens the
time step by factor of 2. This is used namely in more advanced metal models or high numerical aperture focused sources.
THREADS
nthreads
Set total number of threads to be used if we are calculating on CPU (not on GPU). Can be -1 for automated detection of number of cores
and use of all of them which is also default behavior (since GSvit 1.3). Note that on systems using hyperthreading the virtual cores do not help (use half of available cores at maximum). Generally, FDTD
is memory demanding and scaling with number of cores is therefore not linear (memory is the bottleneck), however up to some 8 threads there is
significant speedup. You can test this scaling using ./gsvit test 3N, where N is problem size (e.g. 32 means thread test on 200x200x200 voxels).
As and example, to set the number of threads to 4, write:
THREADS
4
MATMODE_CHECK
0/1
Check material settings and if possible, run computation with smaller memory
allocation - not allocating some of the components and using simplified equations (e.g. for nonmagnetic materials or vacuum). This can save up to half
computation time or memory requirements. A safe variant if something fails is 0 which is
default value (full calculation). This might be necessary for some novel algorithms before they are tested properly, typically algorithms
on GPU where this parameter is not recommended for now.
An example of use is:
MATMODE_CHECK
1
MEDIUM_LINEAR
filename
Use file that consists of binary data representing material, pixel by pixel. File structure
is: xres, yres, zres (binary 32-bit integers) of same size of computation volume,
followed by four 3D fields of 32-bit floats
representing epsilon, sigma, mu and magnetic conductivity.
This approach is ideal for including complex or continuously varying geometries. For simple objects,
use vector data input (MEDIUM_VECTOR), which is much simpler to construct.
MEDIUM_VECTOR
filename
Use file that consists vector material representation, composed from different basic
entities (written in an ASCII file).
Each entity entry is composed by the following information:
- type of entity (integer): 4 - sphere, 7 - cylinder, 8 - parallelepiped, 12 - tetrahedron, 21 - tetrahedal mesh in Tetgen output format, 22 - Gwyddion height field,
107 - cone
- point coordinates in pixel values (doubles): x y z values for each point (single for sphere, two for cylinder, cone
and parallelepiped and four for tetrahedron), plus eventually radius (for sphere, cylinder and cone), or more complex entries (see below) for tetrahedral mesh or Gwyddion height field.
- material type (0: standard material, 1: tabulated material,
2: Drude model (not recommended), 3: CP3 model (not recommended), 4: CP model via Recursive Convolution method, 5: CP model via Auxiliary Differential Equations method,
6: CP model via Piecewise Linear Recursive Convolution method, 10: perfect electric conductor, 99: data from optical database) followed by:
- relative permittivity, relative permeability, electric and magnetic conductivity as double values for material type=0, and 1 (both representing linear material)
- epsilon, omega_p and nu for type=2 (Drude model)
- epsilon, sigma and three sets of a, phi, omega and gamma for type=3 (CP3, in development)
- epsilon, omega, gamma and two sets of a, phi, omega and Gamma for type=4 (CP)
- epsilon, static sigma, omega, gamma and two sets of a, phi, omega and Gamma for type=5 and 6 (ADE and PLRC approach for CP model) (Bug in documentation fixed!)
- nothing for material type=10 (PEC)
- material name for material type=99, e.g. Al2O3
so the entry looks for example as (4 100 100 100 20.5 0 22.13 1 0 0) for sphere with radius 20.5 pixels,
position (100, 100, 100) and relative permittivity of 22.13 (rest or material properties
is same as for vacuum). Material file for two dielectric nonabsorbing spheres and
single absorbing parallelepiped can therefore look like:
4 50 100 100 20.5 0 22.13 1 0 0
4 150 100 100 20.5 0 22.13 1 0 0
8 30 30 160 170 170 170 0 20 1 2000 0
Note that the material data are interpreted succesively, overwriting previous values in the computational volume if there is
an intersection, so you can create relatively complex geometries. Also, if there is MEDIUM_LINEAR,
directive as well in the parameter file, this is performed before MEDIUM_VECTOR data interpretation
(so vector data can overwrite them).
See Example 2 for more details of material parameters use.
For metals, CP model is at present the most suitable approach implemented in GSvit. CP model (in fact Drude + 2 critical points model) is based on this work:
A. Vial, T. Laroche, Appl Phys B (2008) 93: 139-143, giving the following values for different materials
(here already in GSvit material file format including material type (4)):
4 15.833 1.3861e16 4.5841e13 1.0171 -0.93935 6.6327e15 1.6666e15 15.797 1.8087 9.2726e17 2.3716e17 (silver)
4 1.1431 1.3202e16 1.0805e14 0.26698 -1.2371 3.8711e15 4.4642e14 3.0834 -1.0968 4.1684e15 2.3555e15 (gold)
4 1.0000 2.0598e16 2.2876e14 5.2306 -0.51202 2.2694e15 3.2867e14 5.2704 0.42503 2.4668e15 1.7731e15 (aluminium)
4 1.1297 8.8128e15 3.8828e14 33.086 -0.25722 1.7398e15 1.6329e15 1.6592 0.83533 3.7925e15 7.3567e14 (chromium)
Alternatively the same dispersion model can be solved using two other techniques, Auxiliary Differential Equations and Piecewise Linear Recursive Convolution,
whose implementation is both based on K. Chun, H. Kim, H. Kim, Y Chung, PIERS 135, 373-390, 2013 . In the formulas there is a static conductivity term
in addition, which is by many authors treated as zero (absorption is handled by imaginary part of permittivity through the model anyway).
The implementation in GSvit was tested with zero conductivity, giving same reflectivity for flat metal as tabulated values (and same as for Recursive Convolution technique).
In original article by Chun et al. the folloging metals were fitted (presented already with material type and zero conductivity formatted for GSvit use with ADE approach) (Bug in documentation fixed!):
5 0.89583 0 13.8737e15 0.0207332e15 1.3735 -0.504659 7.59914e15 4.28431e15 0.304478 -1.48944 6.15009e15 0.659262e15 (silver)
5 1.11683 0 13.1839e15 0.0207332e15 3.04155 -1.09115 4.20737e15 2.35409e15 0.273221 -1.18299 3.88123e15 0.452005e15 (gold)
5 1.82307 0 13.3846e15 0.163439e15 2.57278 -1.56922e-8 6.65296e15 3.80643e15 0.638294 -1.22019 3.39199e15 0.472389e15 (copper)
If tetrahedral mesh is provided in Tetgen .node and .ele format, the following parameters are given after entity type
(precending material type and material parameters):
filebase attribute_number material_index xshift yshift zshift xmult ymult zmult
Parameter filebase determines the filename of .node and .ele files (filebase.node, filebase.ele)
that are defining the set of tetrahedrons. Attribute_number is used to set which attribute
of the .node and .ele file will be used to assign the appropriate material. Loaded node positions are
multiplied by xmult, ymult, zmult values and then shifted by xshift, yshift, zshift vector. This can be used
to move and scale your data into computation mesh.
If there are no attributes in the .ele file, or there are but attribute_number
is -1, all the tetrahedrons will be treated as the material. If attribute_number is zero or positive and there are attributes
in the .ele file, material_index defines the subset of tetrahedrons (with the same index as the appropriate attribute) that will be used
as material. Using several lines in your vector material definition file using the same filebase
you can assign different materials to your set of tetrahedrons.
As an example a vector material file entry could be following for a cube given by tetrahedral mesh with no attributes (note that no attributes means
no need for attribute number selection), no shift and no scaling of the mesh (mesh is intended for 200x200x200 voxels volume:
21 cube.1 0 0 0 0 0 1 1 1 0 5 1 0 0
Corresponding node file (cube.1.node) would look like this (indentation is not relevant, lines starting with '#' sign before
and after header are skipped):
# my little cube
8 3 0 0
# node list
1 50 50 50
2 151 50 50
3 151 151 50
4 50 151 50
5 50 50 151
6 151 50 151
7 151 151 151
8 50 151 151
Corresponding element file (cube.1.ele) would look like this:
# my little cube
6 4 0
# elements (tetrahedrons), links to corresponding node list
#
1 6 7 1 5
2 1 8 3 4
3 3 7 1 2
4 1 7 6 2
5 1 7 3 8
6 7 8 1 5
If a piece of mesh should not be used, or different materials should be assigned to different
parts of the mesh, use attributes in .ele file as follows:
21 cube.1 0 1 0 0 0 1 1 1 0 4 1 0 0
21 cube.1 0 0 0 0 0 1 1 1 0 5 1 0 0
Here we expect that tetrahedrons have at least single attribute and this attribute (attribute 0) is used
for detection of material. Corresponding element file (cube.1.ele) would now look like this:
# my little cube
6 4 1
# elements (tetrahedrons), links to corresponding node list
#
1 6 7 1 5 1
2 1 8 3 4 0
3 3 7 1 2 1
4 1 7 6 2 0
5 1 7 3 8 1
6 7 8 1 5 0
If Gwyddion height field is provided in Gwyddion format, the following parameters are given after entity type
(precending material type and material parameters):
filename channel mask i j k xoffset yoffset zoffset depth
Parameter filename corresponds to .gwy file that will be loaded (corresponding channel - if you have only
one height field in your file and you haven't done some complex data processing it is probably the channel 0).
If parameter mask is 1, only data under mask are
used for this purposes (see Gwyddion documentation to see what a mask is). If parameter mask is -1, the
inverse of mask is used. Note that real Gwyddion datafield dimensions are used to scale the datafield within the
computational space. Vector
(i j k) in voxel coordinates defines the relative coordinate center in computation space and direction.
(e.g. -1 -1 10 means that the field will be oriented in z normal and its zero coordinate will
be shifted by 10 voxels in z). Parameters xoffset, yoffset and zoffset are in dfield physical coordinates and
are used to further shift the dfield if necessary. If these are zero, top left corner is aligned. Parameter depth (in voxels) defines the
depth to which the material is assigned (with same orientation as the axis).
As an example, the vector material file can look like this for use of channel 2, y axis normal orientation (zero at voxel 50), inverse of mask used,
depth 30 voxels and a slight shift in xy direction:
22 grating.gwy 2 -1 -1 50 -1 1e-7 1e-7 0 30 0 5 1 0 0
Here the computational volume was 200x200x200 voxels and data field had physical dimensions larger than computational volume.
BOUNDARY_ALL
type
Set boundary condition of whole volume to a given type:
- none which means no boundary treatment providing reflection similar to perfect electric conductor,
- liao which means 2nd order absorbing (Liao) providing quite good absorption but not such good as the CPML boundary condition
- cpml which means
convolutional perfectly matched layer (CPML) with parameters:
depth
power
sigma_max
a_max
kappa_max
where sigma_max, alpha and kappa_max are used to generate
coordinate stretching and damping scaled by polynom of power
on CPML region of thickness given by depth.
If sigma_max is set as -1, it will be autmatically
calculated as the optimum value (given by CPML power,
grid spacing and free space impedance).
For computation to be stable, sigma_max and alpha must
be positive and kappa_max must be greater than one.
If kappa_max=1 algorithm is equal to conventional PML
and no coordinate stretching is performed
As an example, to construct a 10 cells thick CPML region
with polynomial scaling with power of 3 and automatically
set optimum sigma_max value use
BOUNDARY_ALL
cpml
10 3 -1 0.03 4
BOUNDARY_X0/BOUNDARY_XN/BOUNDARY_Y0/BOUNDARY_YN/BOUNDARY_Z0/BOUNDARY_ZN
type
Set boundary condition of a given boundary to a given type (types are same as for BOUNDARY_ALL).
Some combinations may lead to instabilities, e.g. corner of cpml and liao region.
MBOUNDARY_X0/MBOUNDARY_XN/MBOUNDARY_Y0/MBOUNDARY_YN/MBOUNDARY_Z0/MBOUNDARY_ZN
type position
Artificial boundary inside material, now only periodic one is supported. Position is in pixel
coordinates. All unset values are set to computational volume boundaries. Only periodic boundary
condition is supported now. To create periodic
boundary condition in x direction ranging from pixel coordinates 10 to 50, you need to set:
MBOUNDARY_X0
periodic 10
MBOUNDARY_XN
periodic 50
Note that periodic boundary condition for total/scattered field formalism incident wave for
angles different from main axes of
computational volume is not supported.
Medium modifiers
MEDIUM_ROUGHEN
radius_peak radius_span iterations probability material_index void_index random_seed
Adds roughness to objects consisting of defined tabulated material (parameter material_index) adding this
material and void or any other material (parameter void_index) to the object randomly.
Roughness is controlled by parameter radius_peak and radius_span (both in voxel coordinates) that
control radius and dispersion of spheres that are added. Parameter probability controls amount of spheres
added in every point. Random seed can be any integer to get same result or -1 to generate seed automatically.
Note that materials (parameter material index) are counted from 1 as they are loaded from vector material
file (so the first material listed there has index of 1). Material with index=0 is vacuum by default
As an example, to add roughness to a sphere (the only object in material file), using 20 iterations
of adding and removing spheres of radius 5+-2 voxels, add this:
MEDIUM_ROUGHEN
5 2 20 0.01 1 0 1
Note that this option is not supported for direct editing by XSvit in this version
MEDIUM_GROW
i0 j0 k0 in jn kn mat_index add_index subsampling nparticles jump_mobility jump_probability random_seed
Simulates growth of tabulated material of given index (parameter add_index) on material with given index (parameter
mat_index). Growth is realized by ballistic deposition using random particle flight and its attachement
that can be followed by some relaxation. Simulation is performed within box defined by (i0 j0 k0 in jn kn)
that can be eventually subsampled (parameter subsampling) to get better control over results. Particles are flying
from box boundaries in random direction unless they leave box or get attached to the material. Materials that
are not listed in this directive have no impact on the process (as they would be vacuum). Jump mobility is
given in voxel units and controls maximum distance allowed for relaxation, jump probability controls
probability of such process (in range of 0-1). Number of particles is typically in range of tens of thousands
at least. Random seed can be any integer to get same result or -1 to generate seed automatically.
As an example to grow material of index=2 (second entry in material file) on material of index=1 (first entry
in material file, with subsampling=2 you can do the following:
MEDIUM_GROW
20 20 20 280 280 280 1 2 2 250e6 4 0.8 1
Note that for high subsampling the process can be slow and memory demanding. Also note that to add material
to the material file without adding any real object (e.g. if you want to grow something that is nowhere else
in your computational geometry) you can add a parallelpiped of zero dimensions.
Note that this option is not supported for direct editing by XSvit in this version
MEDIUM_SMOOTH
unused_integer_parameter
Performs smoothing vector material. Smoothing means locally averaging the optical constants (e.g. permittivity).
This is a very simple way how to reduce staircasing effects. However it is physically reasonable only in
some cases (recalling e.g. effective medium approximation). For metals the procedure is under development and is
not recommended for use in present version and
for PEC it cannot be applied at all.
Field sources
SOURCE_POINT
i j k mode (filename, values)
Point source at position (i j k) using electric and magnetic field intensity
time dependence described in file filename (mode=0) or generating such file for sine or pulsed sine waveform.
If a text file is used, it consists of integer determining total number of values and succesive sets of values consisting of
(step, ex, ey, ez, hx, hy, hz). For "mode=1" the source file is automatically generated (file "tmpsource" in the current
directory) providing a sine wave source; the parameter after "mode" is therefore wavelength
in meters followed by electric field amplitude. Finally, two parameters theta and phi determine the source orientation. For "mode=2" the same happens for sine wave damped by a gaussian envelope,
parameter "mode" is therefore followed by wavelength and by gaussian envelope width,
given in integer steps of the simulation (e.g. 20), and then electric field amplitude. Again, at the end, two parameters theta and phi determine the source orientation.
Theta and phi equal to zero correspond to x-direction electric field point source.
SOURCE_TSF
i_start j_start k_start i_end j_end k_end theta phi psi mode (filename, values)
Plane wave source using total/scattered field formulation. Six integers determine volume that is used
for field propagation, theta and phi determine wave direction and psi its polarisation.
Parameter "mode" distincts different source definition. For "mode=0" a filename is
provided pointing to a text file containing number of values N followed by N pairs of values step number - electric
field value. N needs to be at least the total number of steps in simulation.
For "mode=1" the source file is automatically generated (file "tmpsource" in the current
directory) providing a sine wave source; the parameter after "mode" is therefore wavelength
in meters followed by electric field amplitude. For "mode=2" the same happens for sine wave damped by a gaussian envelope,
parameter "mode" is therefore followed by wavelength and by gaussian envelope width,
given in integer steps of the simulation (e.g. 20), and then electric field amplitude.
Note that in present version the TSF source is in vacuum, material cannot cross it.
Orientation of axes of incoming wave is shown in the following image, table shows
some typical useful values of parameters:
| direction | polarisation | theta [deg] | phi [deg] | psi [deg] | 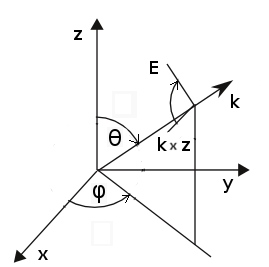 |
|---|
| x axis | y | 90 | 0 | 0 |
| x axis | z | 90 | 0 | 90 |
| y axis | x | 90 | 90 | 0 |
| y axis | z | 90 | 90 | 90 |
| z axis | x | 0 | 0 | 90 |
| z axis | y | 0 | 0 | 0 |
See Example 2 for details of use.
TSF_SKIP
boundary
Specifies boundary that should be excluded from TSF application. Parameter "boundary"
is string denoting which boundary is being set (i0, j0, k0, in, jn, kn). Note that
in principle TSF should be applied on all the boundaries to work properly, but in case
of special materials or boundary conditions some boundary skipping can make sense.
TSF_GAUSSIAN_Z
i_center j_center i_radius j_radius
Enable Gaussian multiplier of the TSF input data providing a Gaussian beam. Working only for TSF source oriented in z direction and for normal incidence.
Beam is centered in i_center, j_center position and has width settable to each direction independently i_radius, j_radius.
Everything is in voxel coordinates.
SOURCE_LTSF
i_start j_start k_start i_end j_end k_end theta phi psi n_materials mat1_pos mat1_epsilon mat1_mu mat1_sigma mat_sigast .... mode (filename, values)
Plane wave source using layered total/scattered field formulation as published by I.R. Capoglu, G.S. Smith: IEEE Transactions on Antennas and Propagation 02/2008, 56:1.
In contrast to SOURCE_TSF, here the material
can be formed by N layers in z-direction (in present implementation only dielectric and non-absorbing). Incident wave however can cross the sample at angle
as well. Layers need to be introduced both in parameters of the source and in material description files (as set MEDIUM_LINEAR or MEDIUM_VECTOR) and the values
of material parameters need to match at LTSF boundary.
First six integers determine volume that is used
for field propagation, theta and phi determine wave direction and psi its polarisation. This is followed by number of interfaces. For every interface its
position in z direction and four material parameters are entered. By default the zero interface starts at
z=0 and has permittivity of vacuum, so it does not need to be entered. Plane wave should start at vacuum. In order to create single free standing film
we therefore need to enter two interfaces, upper and lower as seen in the example below.
Next parameter, "mode", distincts again different source definition (same as for TSF). For "mode=0" a filename is
provided pointing to a text file containing number of values N followed by N pairs of values step number - electric
field value. N needs to be at least the total number of steps in simulation.
For "mode=1" the source file is automatically generated (file "tmpsource" in the current
directory) providing a sine wave source; the parameter after "mode" is therefore wavelength
in meters followed by electric field amplitude. For "mode=2" the same happens for sine wave damped by a gaussian envelope,
parameter "mode" is therefore followed by wavelength and by gaussian envelope width,
given in integer steps of the simulation (e.g. 20), and then electric field amplitude.
As an example to create a plane wave (with a pulse time dependency and polarisation angle 0.3 rad)
incident at angle on a free standing dielectric layer (in 200x200x200 voxels computational volume),
use the following:
SOURCE_LTSF
30 30 30 170 170 170 0.3 0.3 0.3 2 80 3 1 0 0 120 1 1 0 0 2 2e-7 50 5
Note that in this case the material parameters need to match the source, so e.g. using MEDIUM_VECTOR definition we can use this material file:
8 20 20 80 180 180 120 0 3 1 0 0
LTSF_GAUSSIAN
i_center j_center i_radius j_radius
Enable Gaussian multiplier of the LTSF input data providing a Gaussian beam. Now working only for normal incidence.
Beam is centered in i_center, j_center position and has width settable to each direction independently i_radius, j_radius.
Everything is in voxel coordinates.
SOURCE_SF
theta phi psi mode (filename, values)
Plane wave source using pure scattered field formulation, working only for PEC interfaces.
The parameters for source direction and data definition are the same as for TSF source.
Note that SF source is not suported by GPU in present version.
See Example 3 for details of use.
SOURCE_TSFF
i_start j_start k_start i_end j_end k_end thetamax fdist polarisation n m mode (filename, values)
Z direction oriented focused wave source using total/scattered field formulation with multiple plane waves decomposition
according to algorithm published in I.R.Capoglu, A. Taflowe, V. Backman, Optics Express 23 (2008) 19208. Six integers determine volume that is used
for field propagation, thetamax determines maximal angle of incoming light, fdist the focal distance in voxel units.
Polarisation angle of the incoming wave can be controlled by the following parameter.
Integer parameters n and m determine number of plane waves to be integrated (more is better, values around 12, 36 should be already
enough).
Similarily to plane wave source, parameter "mode" distincts different source definition. For "mode=0" a filename is
provided pointing to a text file containing number of values N followed by N pairs of values step number - electric
field value. N needs to be at least the total number of steps in simulation.
For "mode=1" the source file is automatically generated (file "tmpsource" in the current
directory) providing a sine wave source; the parameter after "mode" is therefore wavelength
in meters, followed by electric field amplitude. For "mode=2" the same happens for sine wave damped by a gaussian envelope,
parameter "mode" is therefore followed by wavelength and by gaussian envelope width,
given in integer steps of the simulation (e.g. 20), and finally electric field amplitude.
SOURCE_LTSFF
i_start j_start k_start i_end j_end k_end thetamax fdist polarisation n m n_materials mat1_pos mat1_epsilon mat1_mu mat1_sigma mat_sigast .... mode (filename, values)
Similar focused source to SOURCE_TSFF, however now dielectric material layers arranged in z direction can be crossed, similarly to SOURCE_LTSF.
Note that in contrast to SOURCE_TSFF much more memory is needed for precomputation of the incident field and in present version
you can easily run out of memory for larger systems or larger number of computation steps.
SOURCE_EXT
ipos jpos kpos ijstart jkstart ext_xres ext_yres ext_ifrom ext_jfrom ext_ito ext_jto shift filebase_ex filebase_ey filebase_ez filebase_hx filebase_hy filebase_hz
Uses set of external arrays to inject a source via TSF-like approach. Direction of the source is controlled by ipos, jpos and kpos where only
one of these three parameters should be not -1 similarily to image output. Parameters ijstart and jkstart control placement
of upper left corner of injected source plane. Parameters ext_xres and ext_yres control resolution of the data in external source files
and parameters ext_ifrom, ext_jfrom ext_ito and ext_jto control size and position of the part of external field that is used. Parameters filebase describe
naming of source files (so for filebase_ex="ex" this will be ex_0000, ex_0001 for steps 0 and 1). The format of these files corresponds
to what we get by OUT_PLANE directive in ASCII mode.
Note that this directive is experimental, might be unstable and its syntax might change.
Note that this option is not supported for direct editing by XSvit in this version
SOURCE_WAVELENGTH
min center max
Override automatic wavelength detection mechanism for sources. These (or automatically detected)
values are used when listed material (e.g. silica) is used to pick the optical properties
at right wavelength from the predefined values. Center value is used for materials that are
predefined as lookup table, min and max values are used for materials where we have some
dispersion model available.
Output
Note that in present version every output needs GPU data to be synchronized with CPU, which
can take significant time. For really fast simulations
try to reduce frequency of data outputs.
OUT_FILE
filename.gwy
Set filename that will be used for outputing data. Gwyddion file (*.gwy)
holds all data cross-sections (2d data).
OUT_POINT
Ex/Ey/Ez/Hx/Hy/Hz/All nskip i j k filename
Output values of given component of electric/magnetic field into text
file (filename).
OUT_IMAGE
Ex/Ey/Ez/Hx/Hy/Hz/All/Epsilon/Sigma/Mu/Sigast nskip i j k description
Output image of plane cross-section. Which plane is used is determined
by indices i j k; two of them must be -1. All results are saved to a .gwy file, skipping given
number of steps (e.g. not outputing image in every step unless nskip=1).
Description is a string shown for the channel in Gwyddion data browser.
Note that Gwyddion shows data with top-left corner being center of coordinates,
orientation of axes on what is seen in Gwyddion is show below
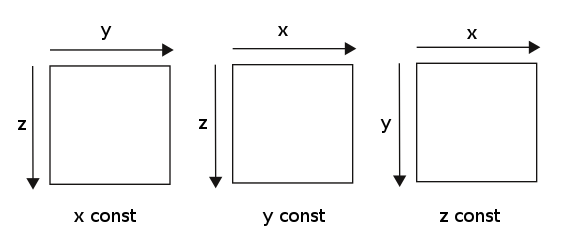
OUT_SUM
component skip i_start j_start k_start i_end j_end k_end epsilon mu sigma sigma* filename
Output sum of electric field intensity (components: ex, ey, ez, all) or absorption
(component: abs) extracted from bounding box and material with given properties (epsilon only used for this).
If epsilon is -1, no control of this parameter is done and whole box is used.
Parameter "skip" controls frequency of file output only (and sync with GPU eventually),
values are calculated at each time step anyway and will be output for intermediate steps as well.
At present this is tested only on CPU.
OUT_SUMTAB
component skip i_start j_start k_start i_end j_end k_end material filename
Output sum of electric field intensity (components: ex, ey, ez, all) or absorption
(component: abs) extracted from bounding box and material with given properties (nk tabulated values
from optical spectra database).
Parameter "skip" controls frequency of file output only (and sync with GPU eventually),
values are calculated at each time step anyway and will be output for intermediate steps as well.
At present this is tested only on CPU.
OUT_VOLUME
Ex/Ey/Ez/Hx/Hy/Hz/All/Epsilon/Sigma/Mu/Sigast/Material/Matmode/Abs skip start stop ascii filename
Output values of given component of electric/magnetic field or material parameters into binary or text
file. Whole computational volume is output. Output starts in "start" step and stops before "stop" step.
If "Abs" component is requested (meaning local absorption), output in every output step is a sum of the present and all the previous outputs
(values in between outputs are not used; this means that to sum absorption from step 100 to 110 you
need to run at least 110 computation steps with volume output skip=1 and volume output start=100, stop=110).
If ASCII file (ascii=1) is output, it has following
structure: xres yres zres channel (and all the data as an array of values separated by space or end of line).
In binary mode the header is omitted, so only arrays are output,
all the array values are doubles (eight bytes), there are no separators. You can read binary output
directly by e.g. Paraview as shown below where a slice of absorption
volume of 100x100x100 voxels is visualised (here you can check also data loading parameters for Paraview).
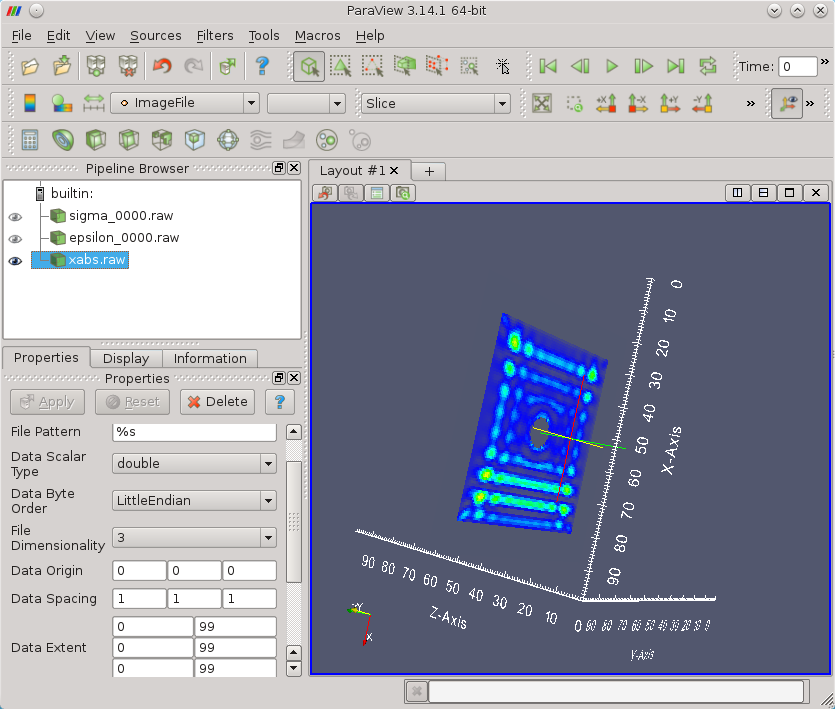
OUT_FORCE
skip i_start j_start k_start i_end j_end k_end filename
Output optical force acting on volume defined by six integers. X, y, and z component
time dependence is output together with its values averaged over source period (source frequency is determined
using FFT). Optical force is calculated using Maxwell stress tensor. No media should cross the
boundary and scaterrer to be evaluated should be placed inside the evaluation volume.
NFFF
i0 j0 k0 i1 j1 k1 sum_from sum_to
Near-to far field calculation boundary definition. Needs to be within the computational
volume. Parameters i0..k1 define the boundary position, sum_from and sum_to define
the range of NFFF data summation when applied (not supported now). See example 3 for details of use.
NFFF_SKIP
boundary i_min/j_min j_min/k_min i_max/j_max j_max/k_max
Specifies area that should be excluded from NFFF calculation. Parameter "boundary"
is string denoting which boundary is being set (i0, j0, k0, in, jn, kn), the next
four numbers describe "upper left" corner and "bottom right" corner of the skipped area.
Multiple calls of NFFF_SKIP can be used to exclude more areas on different boundaries,
however at present only one area on each boundary can be set. Note that
in principle NFFF should be applied on all the boundaries to work properly, but in case
of special materials or boundary conditions some boundary or boundary area skipping can make sense.
NFFF_RAMAHI_POINT
i j k filename
Far field point designed for time values of electric field output. Can be outside
of the computational volume even if it is entered in integer values representing
position in computational volume (therefore values can be e.g. negative). See example 3 for details of use.
NFFF_SPHERICAL_AREA
theta_res phi_res radius theta_from phi_from theta_to phi_to savefile
Creates set of far field points designed for time values of electric field output. Set of (theta_res x phi_res) points
are on a sphere of given radius (int integer i.e. voxel units), theta is declination and phi is azimuth. Parameter savefile
determines whether to save all the farfield point dependencies or just put the total output into general output
file (in Gwyddion format). As an example, considering a surface with normal oriented in z direction (negative),
to cover whole half-sphere above sample for reflection scattering simulation by
set of 20x20 points on sphere with radius of 1000 voxels, and not to save intermediate files, write
NFFF_SPHERICAL_AREA
20 20 1000 90 0 180 360 0
Note that this option is not supported for direct editing by XSvit in this version, however it can be still visualised.
NFFF_PLANAR_AREA
ares bres afrom bfrom ato bto orientation distance savefile
Creates set of far field points designed for time values of electric field output. Set of (a x b) points
are on a plane oriented with normal to direction in x, y or z (depending on "orientation parameter" ranging from 0 (x) to 2 (z).
Distance from computation center can be set, in integer i.e. voxel units. Plane offset in "a" and "b" direction and its
span (that does not need to match voxel spacing) can be controlled by parameters "afrom", "bfrom", "ato", "bto".
Parameter savefile
determines whether to save all the farfield point dependencies or just put the total output into general output
file (in Gwyddion format).
Note that this option is not supported for direct editing by XSvit in this version, however it can be still visualised.
PERIODIC_NFFF
i0 j0 k0 i1 j1 k1 per_ifrom per_jfrom per_ito per_jto
Periodic near-to far field calculation boundary definition. Suitable namely for use with periodic
boundary conditions of same dimensions. Uses only z planes (given by integer k0, k1) but repeats it as many times as given by integer
span (per_ifrom, per_jfrom, per_ito, per_jto). Using periodic repeating values 0 0 1 1 gives the same
result as conventional NFFF with skipped all the boundaries except single z plane. Values of periodic repeating can be
negative, so e.g. values -1 -1 2 2 will produce result of 3x3 copies of calculated fields centered at the originally computed one.
PERIODIC_NFFF_SKIP
boundary i_min/j_min i_max/j_max
Specifies area that should be excluded from periodic NFFF calculation. Parameter "boundary"
is string denoting which boundary is being set (k0 or kn as no other boundaries are used in periodic NFFF), the next
four numbers describe "upper left" corner and "bottom right" corner of the skipped area.
Note that
in principle NFFF should be applied on all the boundaries to work properly, but in case
of special materials or boundary conditions some boundary or boundary area skipping can make sense.
PERIODIC_NFFF_POSTPROCESS
yes/no start
Speedup of perodic NFFF by accumulation of the boundary values during computation followed by their
processing at the end. First parameter requests the algorithm (0 by default not use it, 1 to use it),
second parameter can be used to reduce memory needs skipping integration at beginning of the computation
when field is not developed enough anyway. Still can be very memory demaning for longer calculations.
PERIODIC_NFFF_RAMAHI_POINT
i j k filename
Far field point designed for time values of electric field output in periodic NFFF. Can be outside
of the computational volume even if it is entered in integer values representing
position in computational volume (therefore values can be e.g. negative).
PERIODIC_NFFF_SPHERICAL_AREA
theta_res phi_res radius theta_from phi_from theta_to phi_to savefile
Creates set of far field points designed for time values of electric field output using periodic NFFF. Set of (theta_res x phi_res) points
are on a sphere of given radius (int integer i.e. voxel units), theta is declination and phi is azimuth. Parameter savefile
determines whether to save all the farfield point dependencies or just put the total output power into general output
file (in Gwyddion format). As an example, considering a surface with normal oriented in z direction (negative),
to cover whole half-sphere above sample for reflection scattering simulation by
set of 20x20 points on sphere with radius of 1000 voxels, and not to save intermediate files, write
PERIODIC_NFFF_SPHERICAL_AREA
20 20 1000 90 0 180 360 0
Note that this option is not supported for direct editing by XSvit in this version, however it can be still visualised.
Graphics card use
GPU
ngpus
Set number of graphics cards to be used (indexed as CUDA indexes them).
If this command is not used or set to zero, calculation is performed
on PC processor. See example 1 for details of use. Note that at present
version use of multiple GPUs together is implemented, but untested.
UGPU
number
Set GPU with given index to be used for calculations (indexed as CUDA indexes them).
This can be used for example to run several different calculations on different cards.
See example 1 for details of use.
GPU_QUERY
0/1
Search for available GPUs, detecting and printing their capabilities prior to using any of them (default 1, which means true).
Skipping this step can prevent large delays on some supercomputing systems with very large number of boards
and PCI buses, however it is not necessary nor recommended for use on a standard PC.
General commands
VERBOSE
0-4
Set text output (step by step), from full (4) to silent mode (0).
Examples
There are several examples distributed together with the application.
to test different software algorithms. Please note that if you have installed the Gsvit package into the standard location
(e.g. /usr/share/gsvit/tests on Linux), the software runned as ordinary user won't have permissions to write the results.
Therefore, we recommend to copy the respective test directory to the home directory owned by the user and run the computation from this location.
Basic program operation
Total/scattered field source, PEC sphere
Scattered field source, PEC sphere
Near-to-far field transformation
CPML boundary condition
Periodic boundary conditions
Note that some more examples together with their setup are in GUI (XSvit) documentation.
(c) Petr Klapetek, 2013
| 